Implementing a real-time HIV molecular epidemiology platform
Simon Frost, Mukarram Hossain, Art Poon, Sergei Kosakovsky Pond, Steven Weaver, Erik Volz, Trevor Bedford, Richard Neher, Sten Vermund, Timothy Sterling, Ann Dennis, Marcia Kalish
Introduction
- Pathogen sequence data contain information about their evolutionary past
- Timing and dynamics of spread of epidemics
- High density sampling and rapid sequencing technologies allow us to examine current epidemiological dynamics
- Need to have the appropriate infrastructure
- When combined with models, we can assess the impact of interventions
- Changes in incidence/population structure may be reflected in the sequence data
Real-time molecular epidemiology
- With increased availability, decreased cost, and higher throughput of sequence data, there is increased interest in (near) real-time molecular epidemiology of pathogens
- Needs
- Samples
- Sequencing
- Algorithms
- Platforms
Platforms
- Disease Bioportal (UC Davis)
- Originally for FMDV, now includes influenza A
- Microreact (David Aanensen, Imperial/Sanger)
- Predominantly bacterial pathogens
- nextflu/nextstrain (Trevor Bedford/Richard Neher)
- Originally influenza
- Extended to Ebola, Zika, and other acute viruses
- HIV-trace (Sergei Pond/Steven Weaver/Joel Wertheim)
- HIV, but just for clustering sequences
- Epimatics (Art Poon)
- Not open source
Real-time surveillance of HIV
- At first glance, HIV does not stand out as a good candidate for real time surveillance
- Chronic infection, with diagnosis long after infection
- Typically does not cause outbreaks

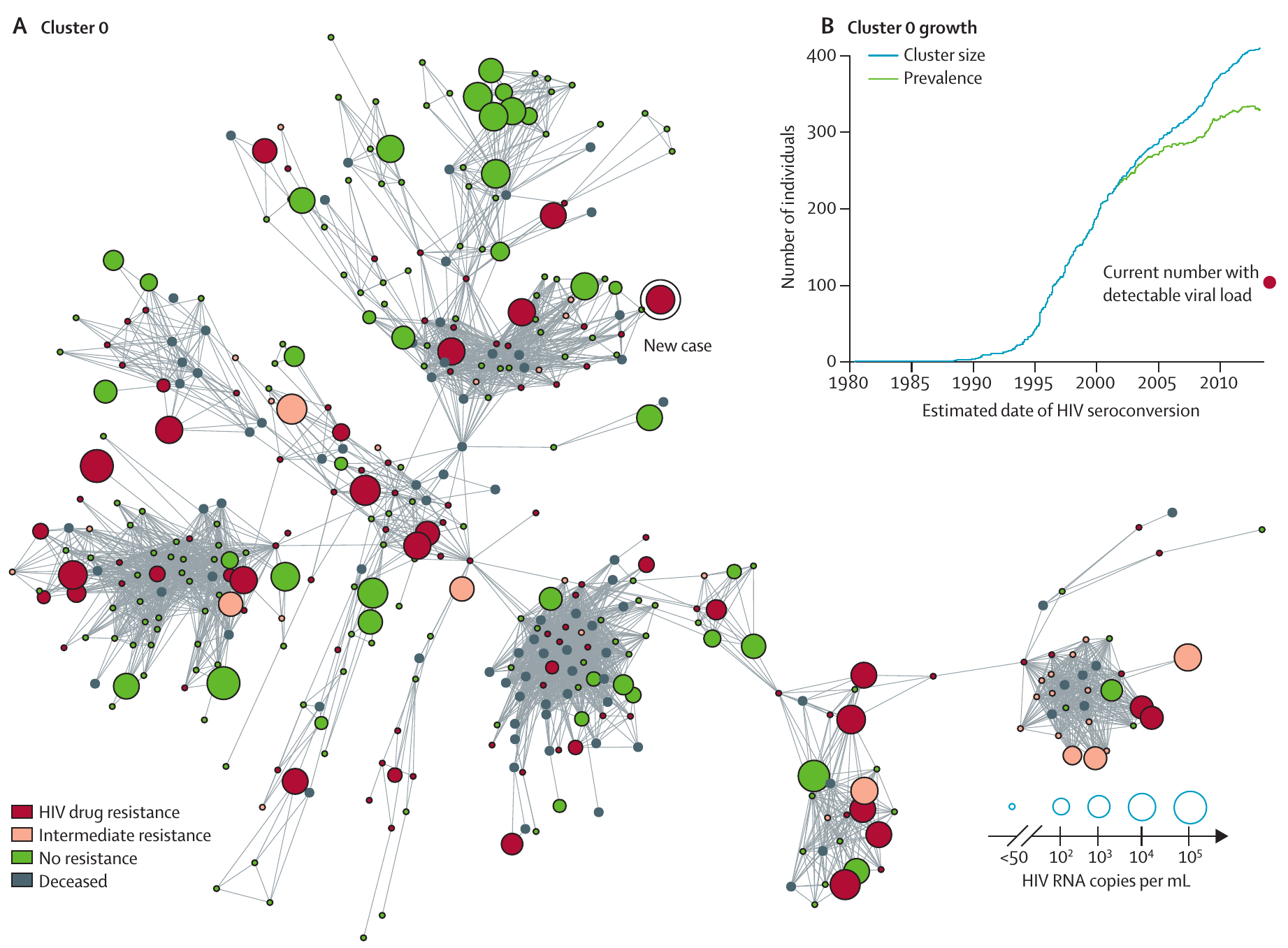
Challenges in implementation
- Most molecular epidemiological analyses are done manually
- Lack of reproducibility
- Labour-intensive
- Data governance issues favour a decentralised system
- Cannot send data ‘out’
- Have to implement all tools within the same system
- Databases may be large
- Need to have fast algorithms
- ‘Online’ approaches to facilitate real-time analyses
- Data are complex
- Need to offer reports, visualisations, etc..
Why not nextstrain?
- Pros:
- Winner of the NIH/Wellcome Trust Open Science Prize
- Cons:
- Linked to specific database platform
- Runs analyses in batch, rather than ‘online’
- Light on data processing
- Moving target
- Enter nextHIV…
nextHIV
- Platform for real-time HIV surveillance
- Sequence data
- Metadata
- Allows multiple sequences per individual
- Takes ideas and the ‘good bits’ from other platforms
- Benefits of open source
- In collaboration with Trevor, Richard, Sergei, etc.
- Emphasis is on speed and to deploy locally
Application
- Since 2001, the Vanderbilt Comprehensive Care Clinic (VCCC) has been generating HIV sequence data for clinical care
- Most patients included in the North American AIDS Cohort Collaboration on Research and Design
- Validated clinical endpoints, demographics, treatment data and regimens
- 2,915 individuals, 4,727 sequences with demographic data
- 773 individuals, 1505 sequences without demographics
- Over 170,000 PR/RT sequences from the US in public databases
- Very few come from Tennessee
nextHIV workflow
- Imports data into a database
- Aligns it against a standard HIV reference strain
- Generates drug resistance interpretations
- Compares sequences against a large reference database
- Infers subtypes
- Generates clusters of related sequences
- Generates phylogenies
- Generates interactive reports
Alignment
- Multiple sequence alignment can be slow for large numbers of sequences
- Map to the reference HXB2 using the Python library BioExt (Hepler/Weaver/Pond)
- Fast (and parallelised)
- Codon-aware
- Allows us to use HXB2 numbering
- Aligns over 20 sequences per second on a standard laptop
Drug resistance
- We employ the Stanford drug resistance scoring scheme
- Rather than use a web service, the scheme is implemented locally
- ‘Naive’ individuals determined using regimen data
Transmitted NNRTI resistance
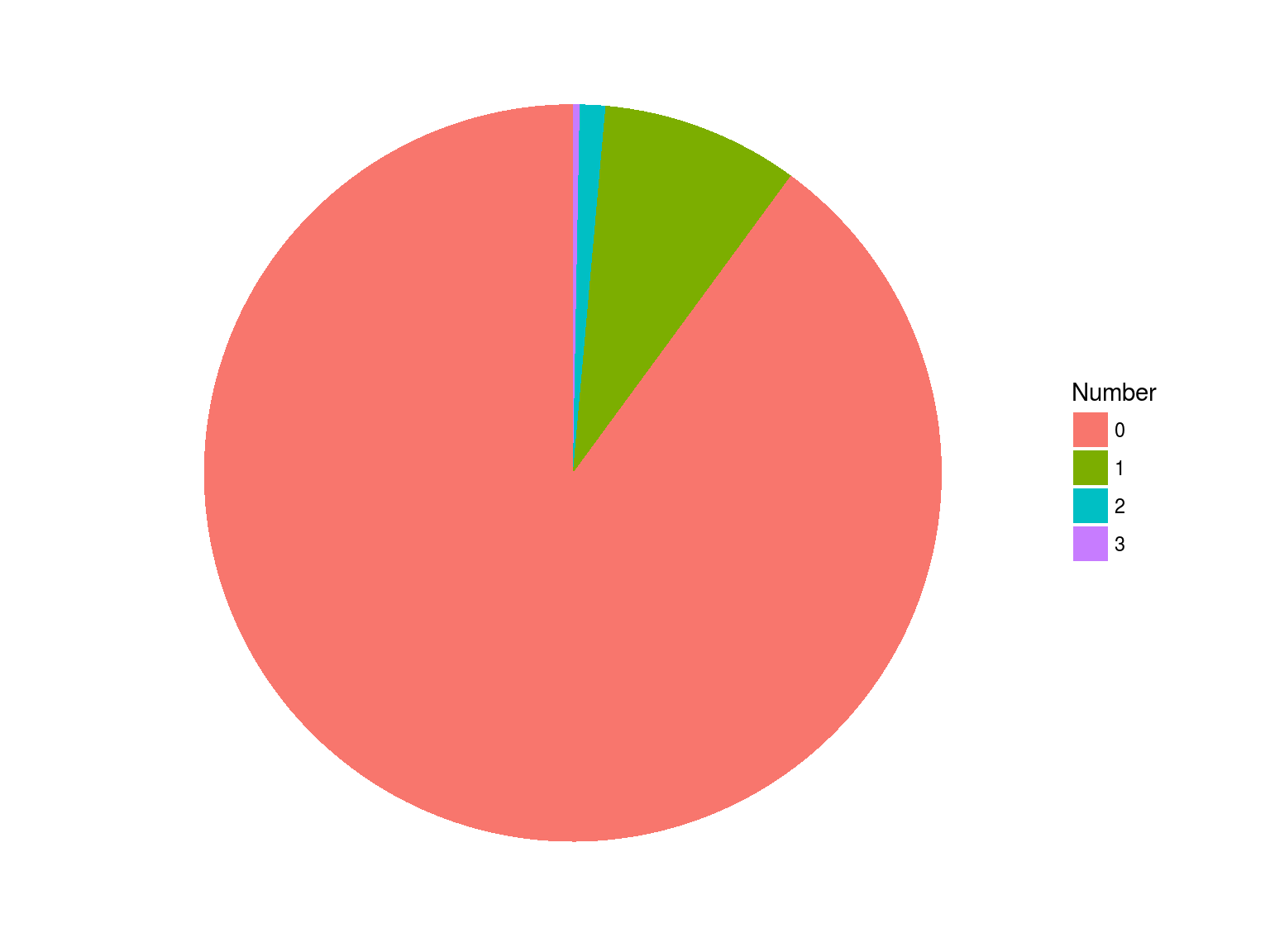
Distance calculations
- For clustering we use an implementation of the TN93 distance by Sergei
- Part of HIV-TRACE
- Refactored into a Python/C library
- Copes with ambiguous nucleotides systematically
- Compute all pairwise distances between cohort sequences
- Identify closest matches from a reference alignment
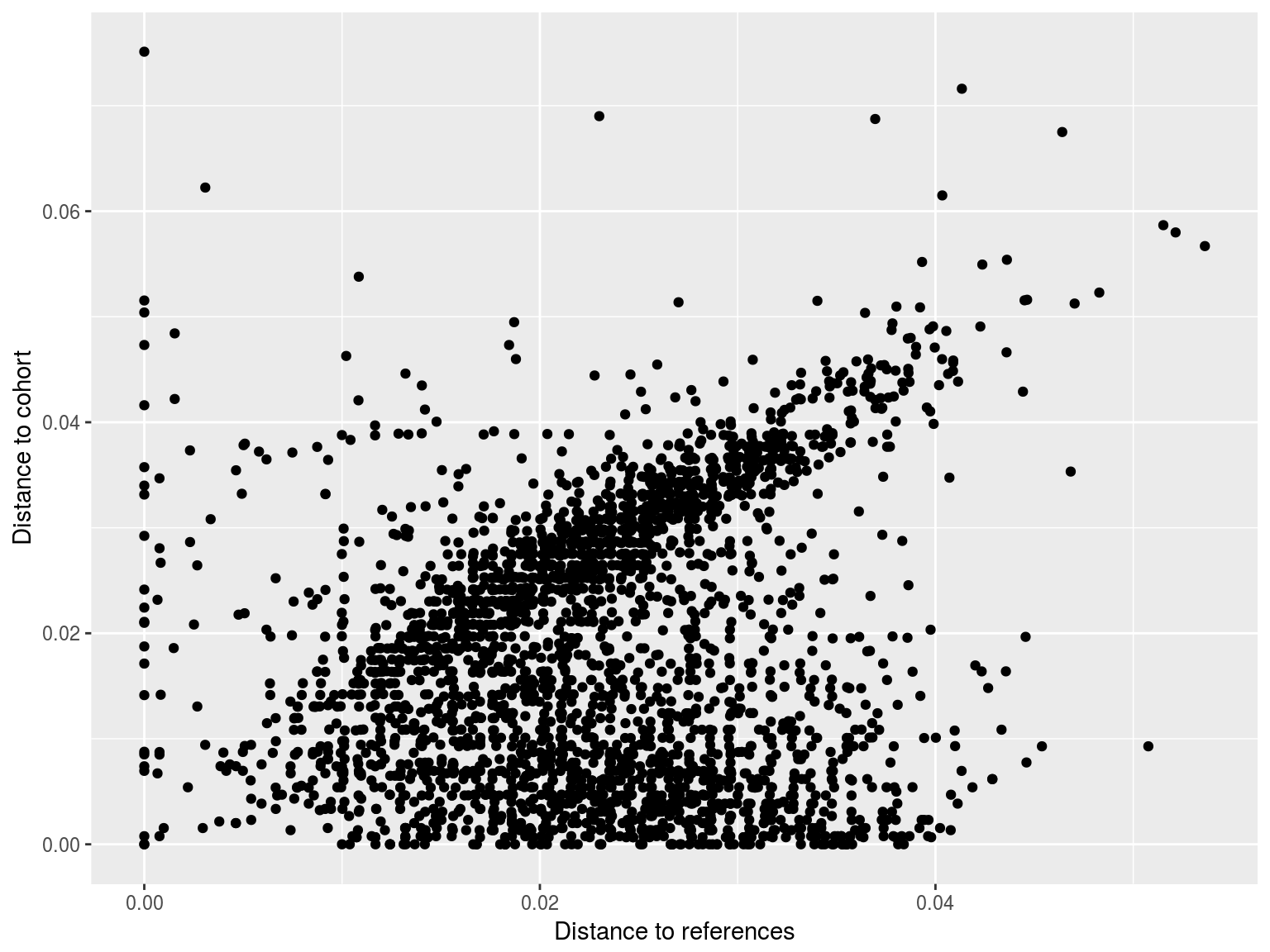
Clustering
- To help overcome some of the problems with analysing large sequence datasets, many HIV studies focus on clusters of similar sequences
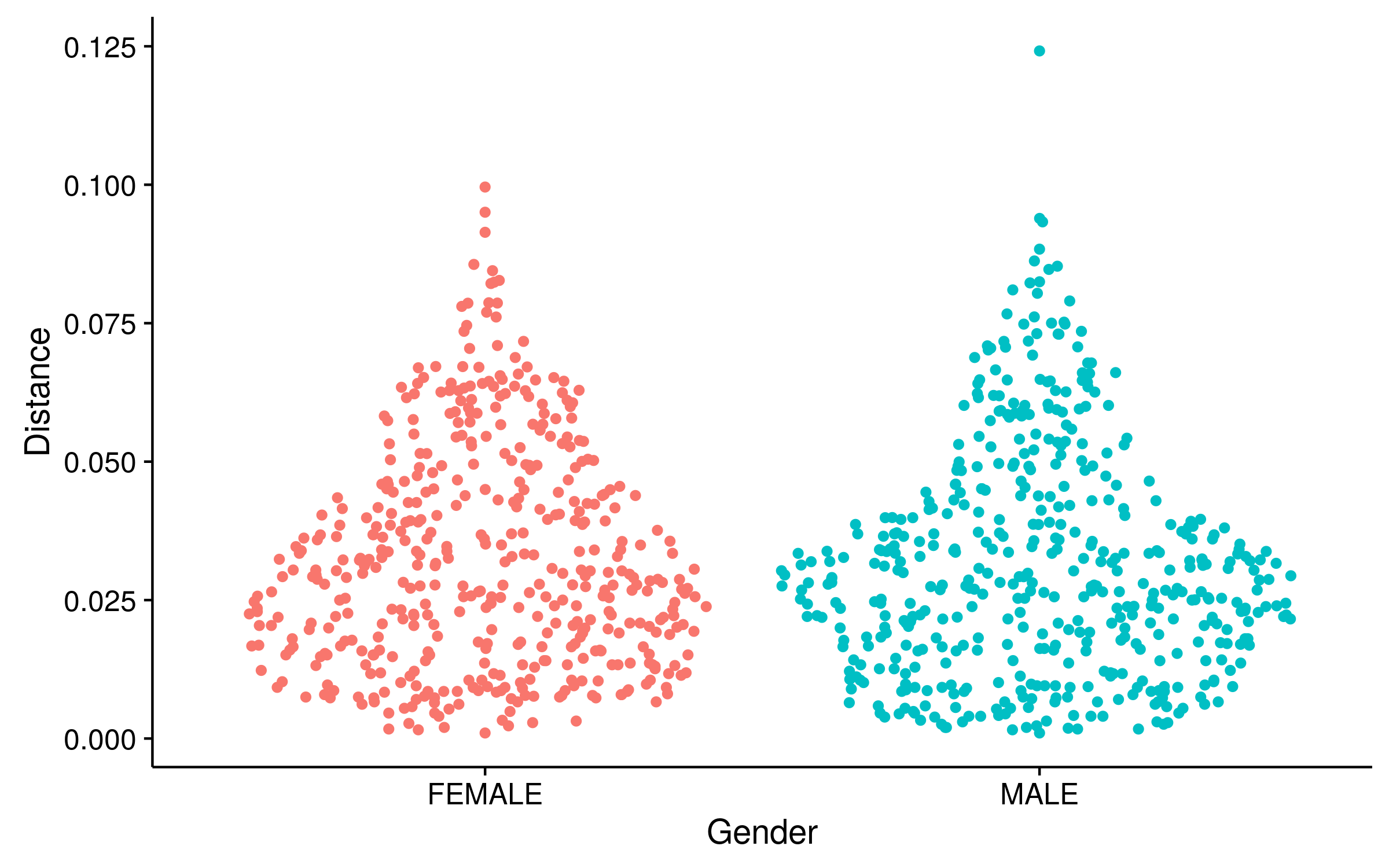
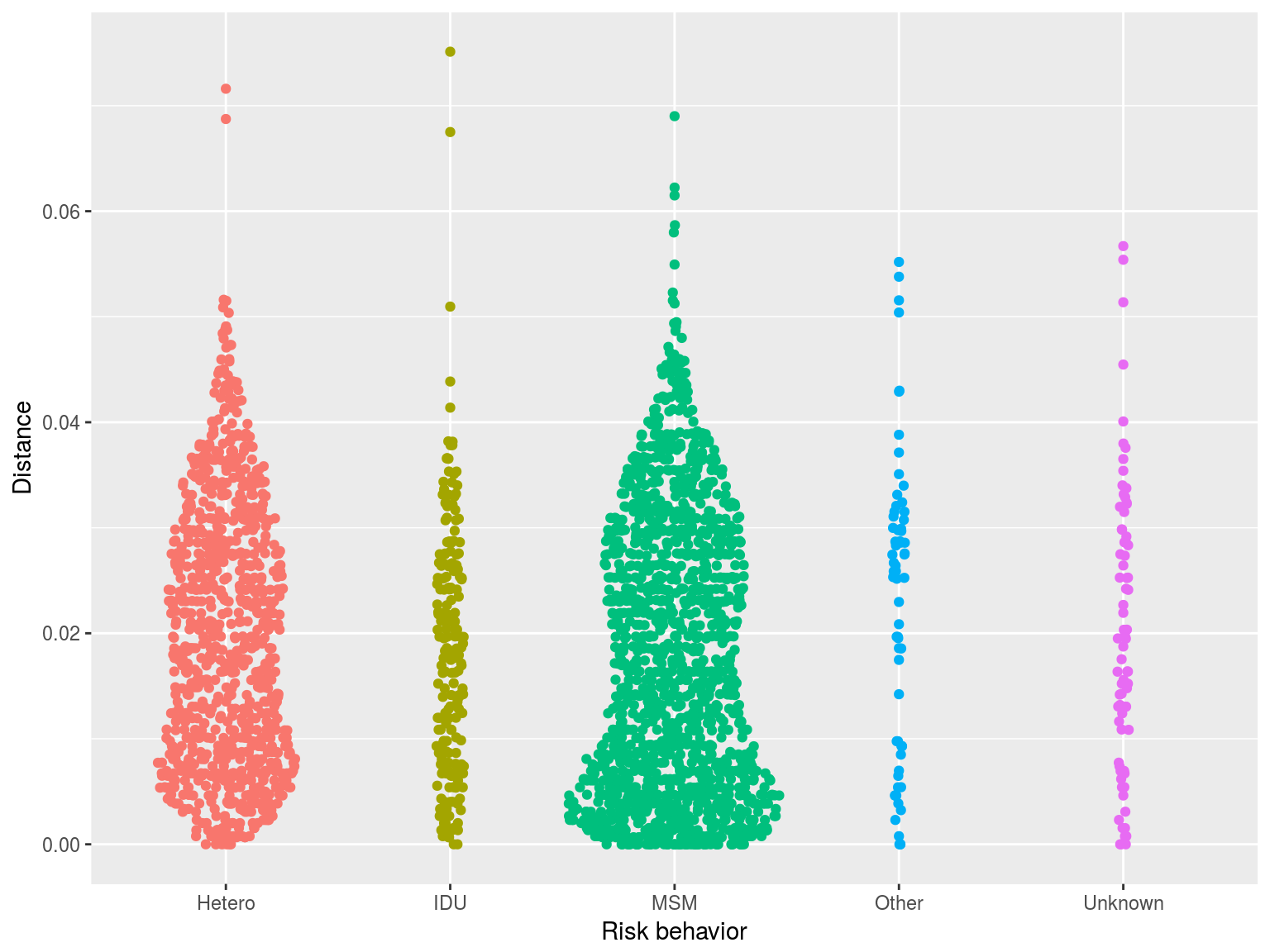
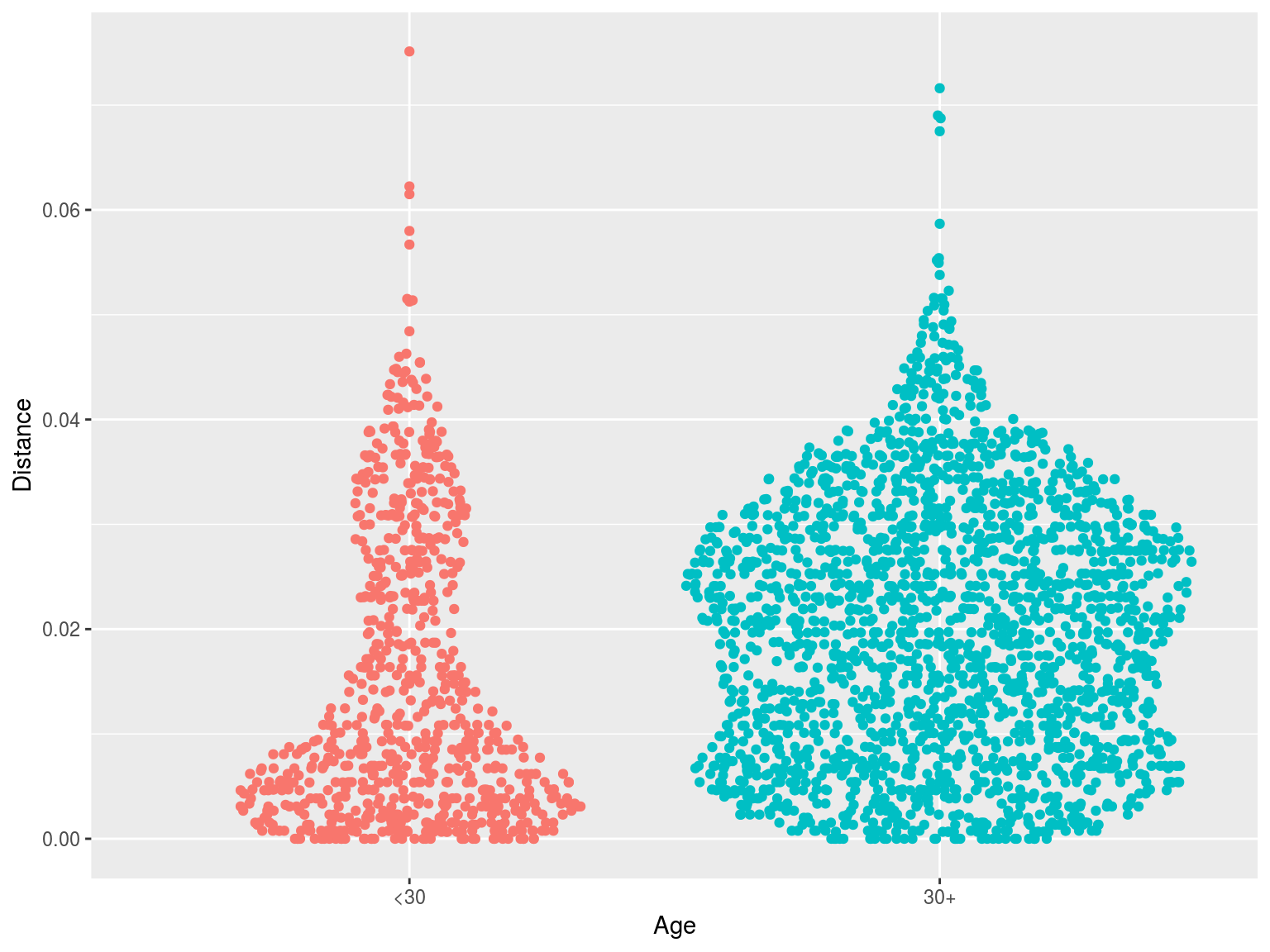
- One simple approach we have used is to calculate the distance to the nearest sequence
- Closer sequences = more clustering
- This approach does not involve choosing a threshold
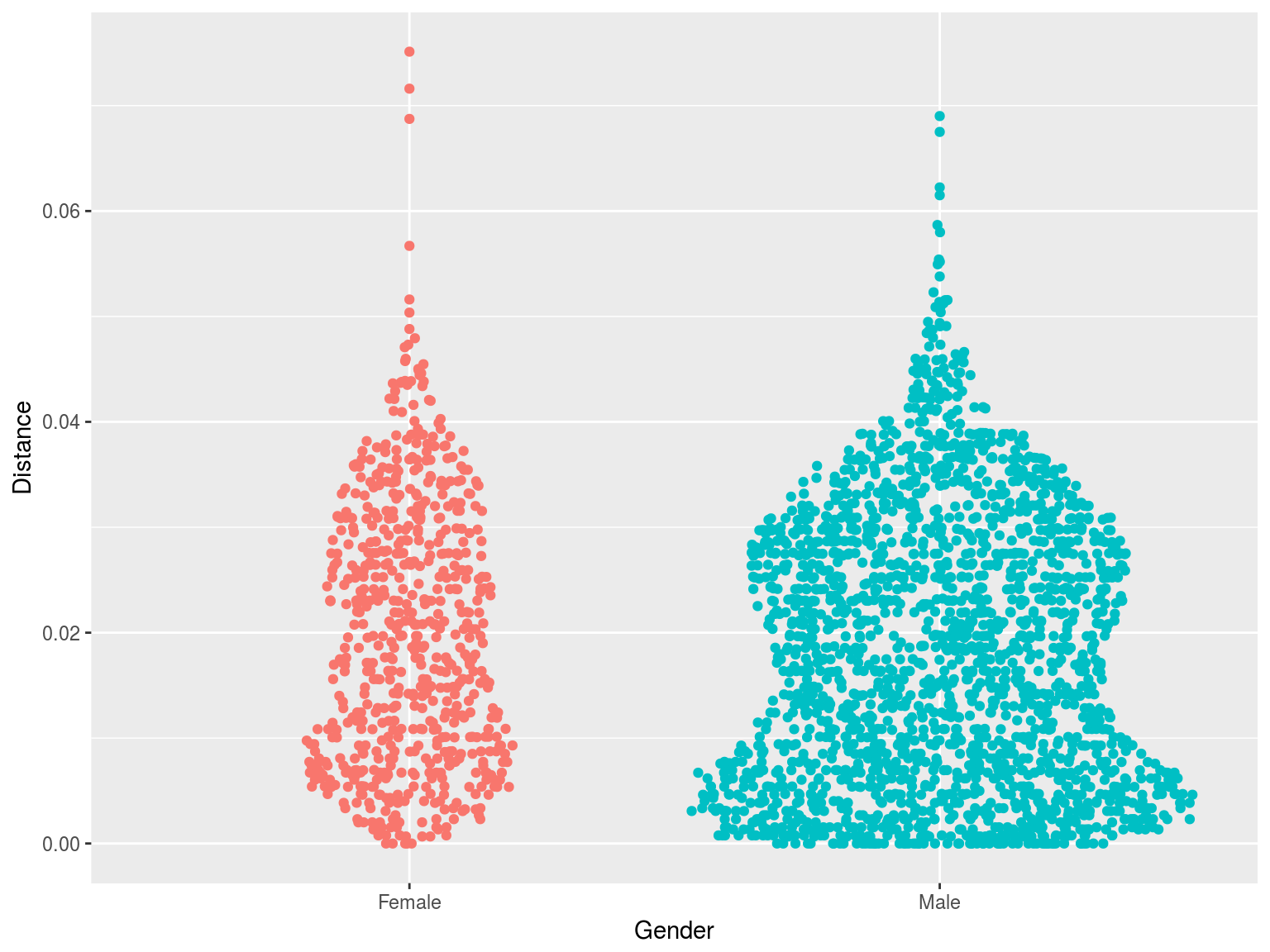
Clustering
Clustering by gender
Clustering in PANGEA village simulations
Clustering by risk group
Clustering by age
Subtype assignment
- Whilst SCUEAL and Rega v3 are arguably the standard, we would like something much faster
- Candidates:
- COMET
- Uses a prediction by partial matching approach
- Good performance
- Not open source
- STAR
- Uses a position-specific scoring matrix
- Not actively maintained
- COMET
nextHIV subtying pipeline
- Subtype of closest (TN93) reference
- pyCOMET
- An open source implementation of COMET in Python
- Alignment free
- PSSM
- An open source PSSM method along the lines of STAR
- Requires alignment
Non-B subtypes
Phylogenetic reconstruction
- We use RAxML and IQTREE for phylogenetic reconstruction
- Both can add single sequences to an existing tree
- For molecular epidemiological purposes, we would like time-calibrated trees
- We employ
treedater(Volz)- Fast, relaxed clock approach
- We employ
treedater performance

Phylogenetic processing
- In many settings, we may have many sequences from a given subtype
- Results in phylogenies that are difficult to interpret
- We consider smaller subtrees:
- Slice the phylogeny at a given time
- Carve into subtrees that are monophyletic with respect to the reference sequences
Subtype B
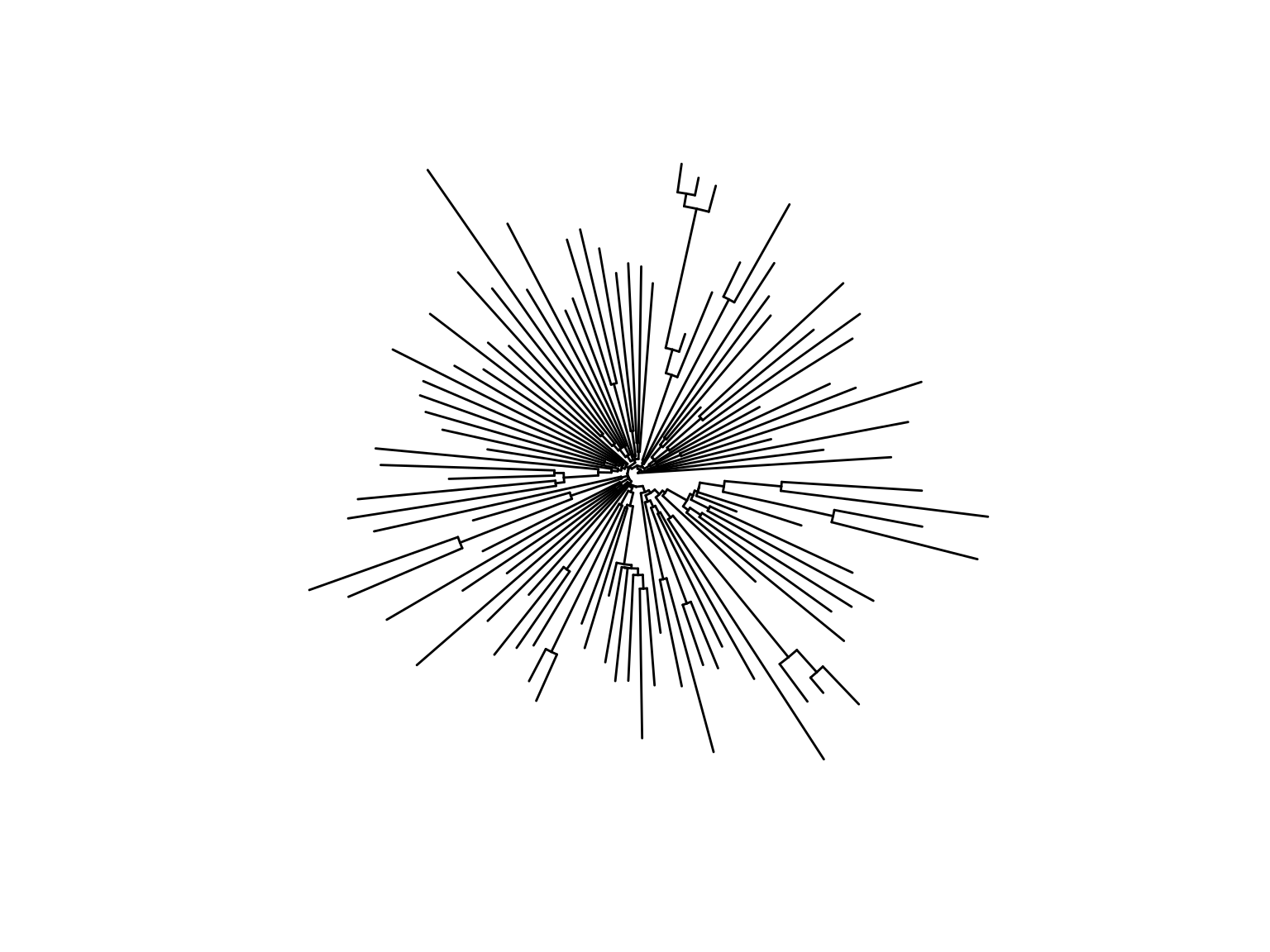
Parsimonious introductions

Cluster distributions
- 2783 subtype B sequences from unique patients
- 931 clusters
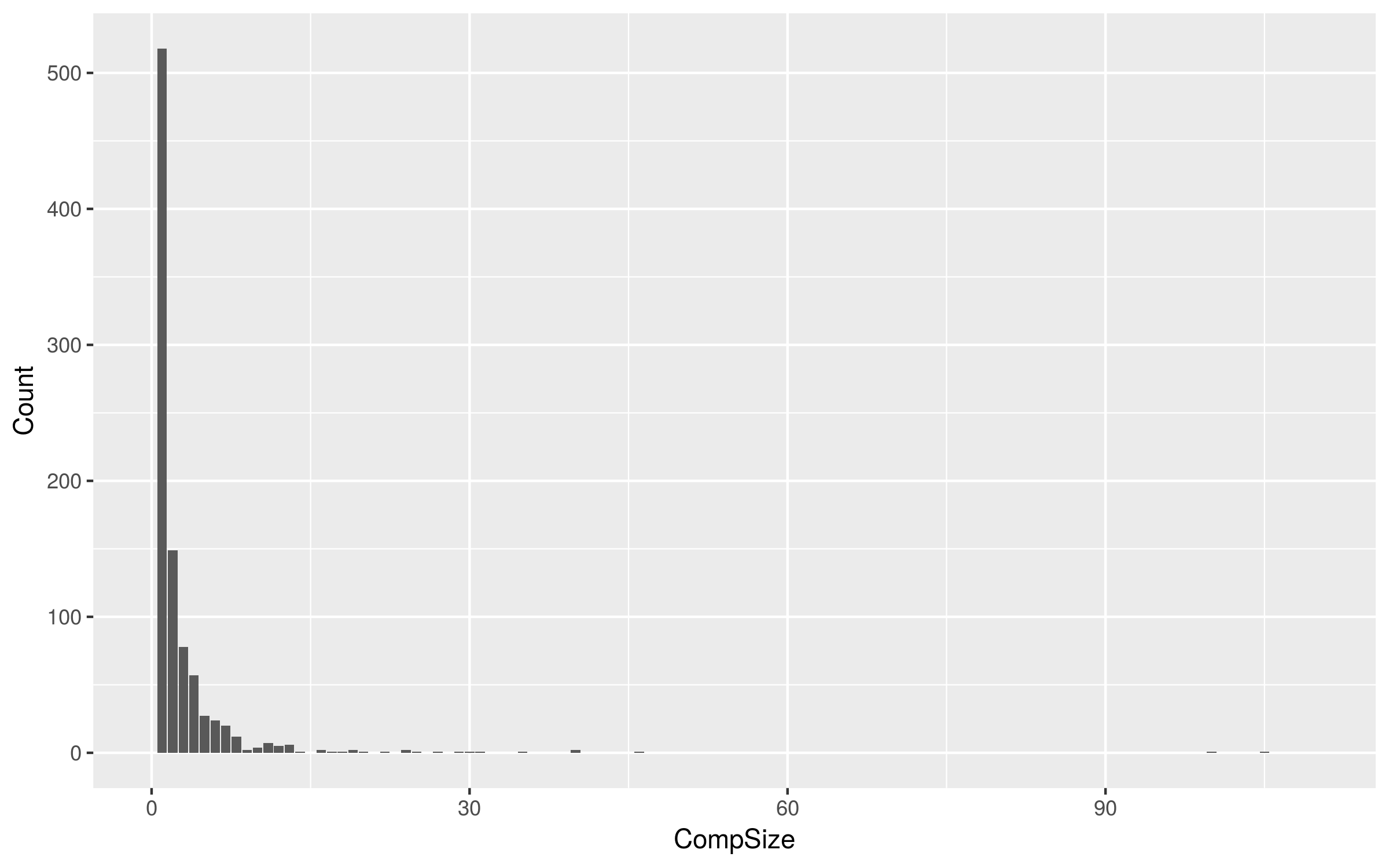
Largest monophyletic cluster
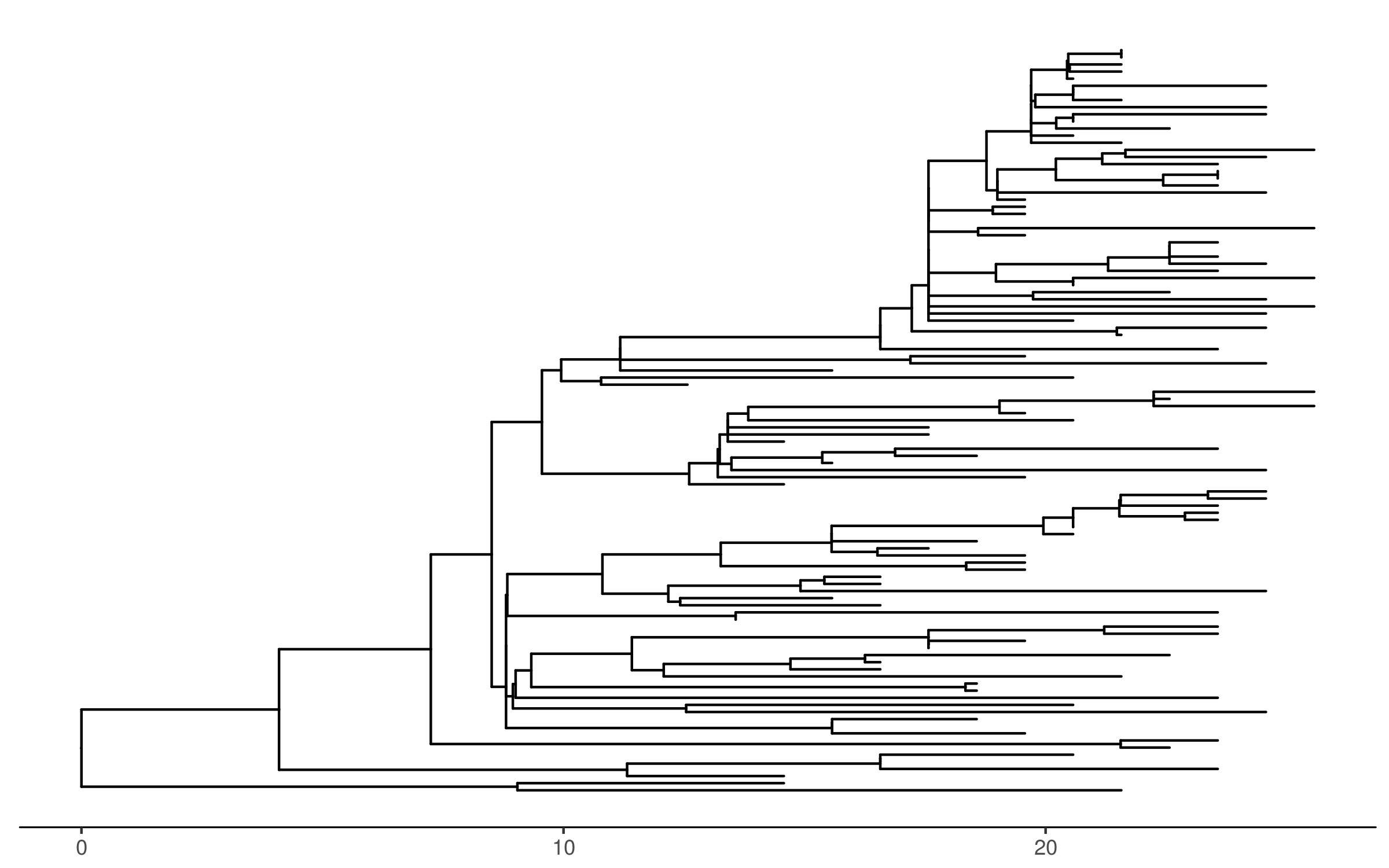
Characteristics of largest cluster
- Young
- Median 26 yo
- African American
- 90/105
- Male, MSM
- 91/105 MSM
- Geographically dispersed
- Majority come from 3 ZIP3 codes
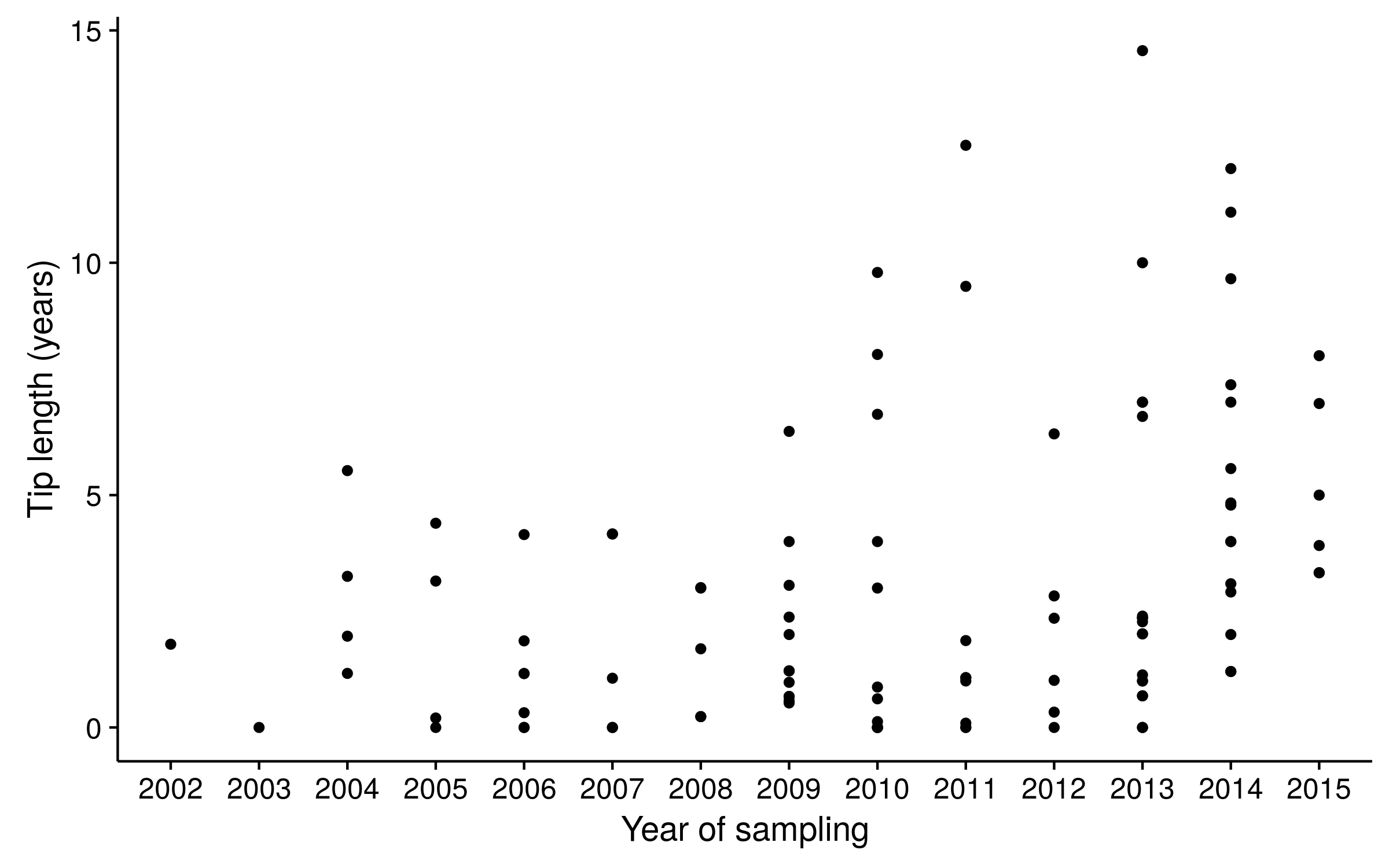
Tip lengths
Reporting
- Considerations
- Interactivity is desirable
- Self-contained (detached from source data)
- Easily customiseable
- R Markdown document
- Easy to include interactive ‘widgets’
Configuration
- Configuration is performed with a simply formatted text file
sequence:
table: 'sequences'
column: 'SEQUENCE'
name: 'SEQNAME'
pid: 'CFAR_PID'
ordering: 'AGE_AT_SEQUENCE'
collection_date: 'YEAR_OF_SEQUENCE'
startpos: 0
endpos: 1497
processed_table: 'processed_sequences'
processed_name: 'SEQUENCE_ALIGNED'Implementation
- Open source (Python/R)
- Can use different database backends to store data
- Is ‘containerised’ to allow running on Windows, Mac, Linux servers
- Currently Docker container
- Many analyses run in parallel (if on a multicore machine)
Work in progress
- NGS/WGS
- Geographical analyses
- Standards for interoperability?
Summary
- We have developed a platform for real-time HIV surveillance
- Could be used for other viruses too
- Alongside algorithmic development
- Time calibrated trees (Erik)
- Subtyping (Mukarram)
- Counting introductions

